from dgrec.example_data import get_example_data_dirlibrary_size
Functions to estimate the total library size (number of unique genotypes) from sampling data. Fits a hybrid power-law distribution model to genotype frequency data and extrapolates to estimate unobserved genotypes.
estimate_library_size
def estimate_library_size(
observed_counts:list, G:int, # total number of possible unique genotypes
N:int, # number of cells
verbose:bool=False
)->dict:
Estimates the expected number of unique genotypes sampled (K_N).
This function uses a hybrid model: 1. The “head” (high-frequency genotypes) uses empirical probabilities. 2. The “tail” (low-frequency and unobserved genotypes) is modeled with a power-law (Zipf) distribution.
Args: observed_counts (list[int]): A list of UMI counts for each observed unique genotype. G (int): The total number of possible unique genotypes. N (int): The number of cells in the culture to sample from.
Returns: dict: A dictionary containing the results: ‘E_KN’ (float): The final estimated E[K_N]. ‘B’ (int): The estimated breakpoint. ‘alpha’ (float): The estimated power-law exponent. ‘E_head’ (float): The contribution to E[K_N] from the head. ‘E_tail’ (float): The contribution to E[K_N] from the tail.
find_best_breakpoint
def find_best_breakpoint(
counts:ndarray
)->tuple:
Finds the optimal breakpoint B for a hybrid power-law distribution.
This function iterates through possible breakpoints and performs a linear regression on the log-log plot of the tail of the distribution (rank vs. probability). The optimal breakpoint is chosen as the one that maximizes the R-squared value of the linear fit, indicating the point where the power-law behavior is most pronounced.
Args: counts (np.ndarray): An array of UMI counts for each observed genotype, sorted in descending order.
Returns: tuple[int, float, float]: A tuple containing: - best_b (int): The estimated optimal breakpoint B. - best_alpha (float): The estimated power-law exponent alpha for the tail. - best_intercept (float): The intercept from the log-log linear regression.
data_path=get_example_data_dir()
gen_list=parse_genotypes(os.path.join(data_path,"sacB_genotypes.csv"))
read_ref_file="sacB_ref.fasta"
ref=next(SeqIO.parse(os.path.join(data_path,read_ref_file),"fasta"))
ref_seq=str(ref.seq)
#showing a few example lines
for g,n in gen_list[1:200:20]:
print(n,"\t",g)279 A91G
28 A68C
15 A72G,A79T,A91T
10 A61G,A72G
6 A61G,A68G
6 A68G,A76G,A91G
5 A61T,A79G
4 A86T
4 A72G,A76G,A86G,A91T
3 A61T,A76G,A91G# Demo: find_best_breakpoint
counts_demo = np.sort(np.array([c for g,c in gen_list], dtype=np.float64))[::-1]
counts_demo = np.trim_zeros(counts_demo)
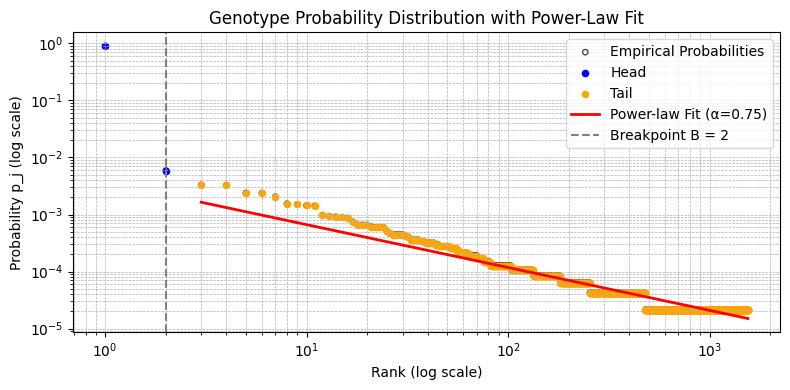
B, alpha, intercept = find_best_breakpoint(counts_demo)
print(f"Breakpoint B={B}, alpha={alpha:.4f}, intercept={intercept:.4f}")Breakpoint B=2, alpha=0.7513, intercept=-5.5858counts = [c for g,c in gen_list]
results = estimate_library_size(counts, G=4**20, N=10**9)
results{'library_size': 89740015,
'B': 2,
'alpha': 0.7512643654095239,
'C': 2.3011837490037884e-05,
'intercept': -5.585815841202696,
'E_head': 2.0,
'E_tail': 89740013.23215128}plot_distribution
def plot_distribution(
counts:ndarray, results:dict
):
Plots the probability distribution on a log-log scale.
Args: counts (np.ndarray): Sorted array of observed genotype counts. results (dict): The dictionary returned by estimate_expected_genotypes.
plot_distribution(counts, results)
Displaying plot...