Usage: dgrec genotypes [OPTIONS] FASTQ REF
Options:
-u, --umi_size INTEGER Number of nucleotides at the beginning of
the read that will be used as the UMI
-q, --quality_threshold INTEGER
threshold value used to filter out reads of
poor average quality
-i, --ignore_pos LIST list of positions that are ignored in the
genotype, e.g. [0,1,149,150]
--match FLOAT match parameter of the aligner
--mismatch FLOAT mismatch parameter of the aligner
--gap_open FLOAT gap_open parameter of the aligner
--gap_extend FLOAT gap_extend parameter of the aligner
-r, --reads_per_umi_thr INTEGER
minimum number of reads required to take a
UMI into account. Using a number >2 enables
to perform error correction for UMIs with
multiple reads
-s, --save_umi_data TEXT path to a csv file to save the details of
the genotypes reads for each UMI. If None
the data isn't saved.
-o, --output TEXT output file path
--help Show this message and exit.dgrec
A Python package for analyzing DGRec (Diversity Generating Retroelement + Recombineering) data
Overview
DGRec is a novel in vivo hypermutation technique that combines two biological systems:
- DGR (Diversity Generating Retroelement): The DGR reverse transcriptase (bRT) + Avd reverse-transcribes a Template Repeat (TR) RNA sequence, introducing errors predominantly at adenine positions.
- Recombineering (CspRecT + mutL*): The single-stranded recombinase CspRecT integrates the mutagenic cDNA into the Variable Repeat (VR) in a target gene, while mutL* prevents mismatch repair from correcting the mutations.
This creates a powerful tool for targeted in vivo diversification in E. coli, where adenine positions within the TR are selectively mutagenized while other bases remain largely unchanged.
The dgrec package provides tools to:
- Call genotypes from amplicon sequencing data (single-end or paired-end), with UMI-based deduplication to correct PCR and sequencing errors
- Visualize mutation profiles at nucleotide and amino acid resolution
Publication: Targeted in vivo hypermutation with DGRec
Documentation: dbikard.github.io/dgrec
Install
We recommend installing dgrec in a dedicated conda environment:
conda create -n dgrec python=3.11
conda activate dgrecInstall ViennaRNA (required for the TR scoring functions):
conda install -c conda-forge -c bioconda viennarnaThen install dgrec:
pip install git+https://github.com/dbikard/dgrec.gitHow to use
Command line interface
Single reads
dgrec genotypes fastq_path reference_path -o genotypes.csvPaired reads
dgrec genotypes_paired fwd_fastq_path rev_fastq_path reference_path --fwd_span 0 150 --rev_span 30 150 -o genotypes.csvUsage: dgrec genotypes_paired [OPTIONS] FASTQ_FWD FASTQ_REV REF
Calls dgrec.genotypes_paired.get_genotypes_paired
Options:
--fwd_span <INTEGER INTEGER>...
Span of the reference sequence read in the
forward orientation format: start end
[required]
--rev_span <INTEGER INTEGER>...
Span of the reference sequence read in the
reverse orientation format: start end
[required]
-p, --require_perfect_pair_agreement
Require perfect pair agreement for genotype
calling (default: True). If
set to False, the forward sequence will be
used in case of disagreement.
-u1, --umi_size_fwd INTEGER Number of nucleotides at the beginning of
the fwd read that will be used as the UMI
(default: 10)
-u2, --umi_size_rev INTEGER Number of nucleotides at the beginning of
the rev read that will be used as the UMI
(default: 0)
-q, --quality_threshold INTEGER
Threshold value used to filter out reads of
poor average quality (default: 30)
-i, --ignore_pos LIST List of positions that are ignored in the
genotype (default: [])
--match FLOAT match parameter of the aligner
--mismatch FLOAT mismatch parameter of the aligner
--gap_open FLOAT gap_open parameter of the aligner
--gap_extend FLOAT gap_extend parameter of the aligner
-r, --reads_per_umi_thr INTEGER
Minimum number of reads required to take a
UMI into account (default: 0).
Using a number >2 enables to perform error
correction for UMIs with multiple reads
-s, --save_umi_data TEXT Path to a csv file to save the details of
the genotypes reads for each UMI. If None
the data isn't saved (default: None)
-n INTEGER Number of reads to use. If None all the
reads are used (default: None)
-o, --output TEXT Output file path
--help Show this message and exit.In python
The package can also be used directly in Python for more flexibility.
Calling genotypes
Load a FASTQ file and a reference sequence, then call genotypes with UMI deduplication. The ignore_pos parameter excludes positions at the edges of the amplicon where sequencing quality is low.
import dgrecfrom Bio import SeqIO
import os
#Getting the path to the fastq file
fastq_file="sacB_example.fastq.gz"
fastq_path=os.path.join(data_path,fastq_file)
#Getting the reference sequence for the amplicon
read_ref_file="sacB_ref.fasta"
ref=next(SeqIO.parse(os.path.join(data_path,read_ref_file),"fasta"))
ref_seq=str(ref.seq)
#Generating a list of genotypes sorted by the number of UMIs that are read for each genotype
gen_list = dgrec.get_genotypes(fastq_path, ref_seq, ignore_pos=[0,1,2,138,139,140,141])
#Printing the top results
for g in gen_list[:20]:
print(f"{g[1]}\t{g[0]}")n reads: 1000
n_reads pass filter: 847
n_reads aligned: 824
Number of UMIs: 814
Median number of reads per UMI: 1.0
Number of genotypes: 123
675
3 C56A
3 A76G
3 A91G
3 A91T
2 C69T
2 T122A
2 A91C
2 A105G
2 C116A
2 T60A
2 T59A
2 A68G
2 T134A
1 A61G,-63T,A76T,A91T
1 A79T,A91G
1 A61G,A72G,A76G,A79T
1 T108A,G127T,G132T
1 A48T,A86G
1 A61T,A68T,A72G,A79C,A91GEach genotype is represented as a comma-separated list of mutations in the format [RefBase][Position][AltBase]. The list is sorted by the number of UMI-deduplicated molecules supporting each genotype. The first entry is typically the wild-type (unmutated) sequence.
Visualizing mutations
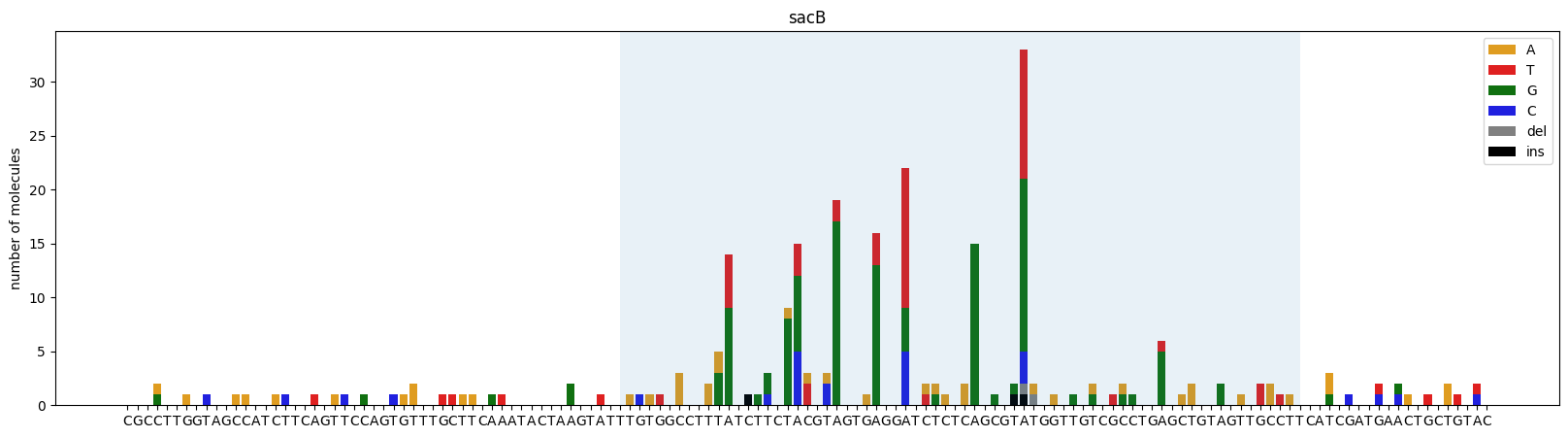
Plot mutation counts at each position. The shaded region indicates the TR (Template Repeat) range where DGRec mutagenesis is active. Note the strong enrichment of mutations at adenine positions within the TR — the hallmark DGRec signature (in this example the TR was designed to have an identical sequence to the VR).
fig = dgrec.plot_mutations(gen_list, ref_seq, sample_name="sacB", TR_range=[50,119])