def optimize_sequence(
original_seq, # Original DNA sequence to optimize.
frame_offset:int=0, # Reading-frame offset (0, 1, or 2) used when grouping codons.
dict_allowed_AAs:NoneType=None, # Dictionary of positions (keys) and AAs (values) where you want to reach all AAs in the list with the codon. If not mentioned, does as before.
Selects for codons which do not reach (by adenine mutation) stop codons. If not possible, allow them anyway.
dict_allowed_AAs_max_min:NoneType=None, # Dictionary of positions (keys) and either you want maximum diversity ('max') or mimimum diversity ('min') at the positions mentionned in dict_allowed_AAs. Diversity = number of AAs reachable by adenine mutations (already removed codons reaching stop codons).
If not mentioned, any sequence that fullfills dict_allowed_AAs[i] is accepted.
If there is no list of accessible AAs for dict_allowed_AAs[i] but dict_allowed_AAs_min_max[i]=='max', it puts an AAC here and forbids the algorithm to change it.
CHANGES:int=6, # Maximum number of codon substitutions allowed (on top of the AAs requirements from the previous argument).
freq_min:float=0.2, # Lowest usage frequency acceptable.
N:int=1, # Number of putative TR to output.
forbidden_positions:list=[], # Nucleotide positions that must not be modified.
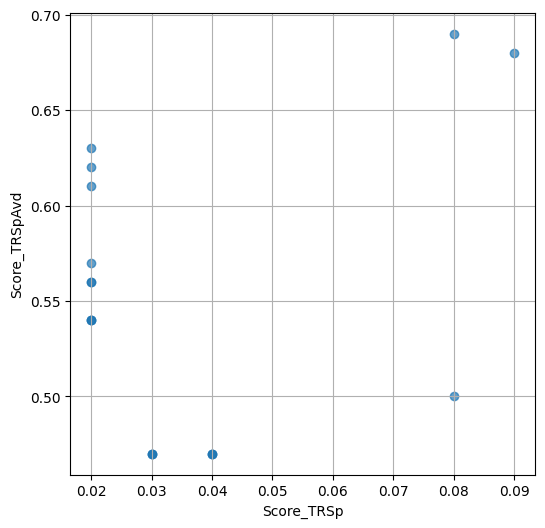
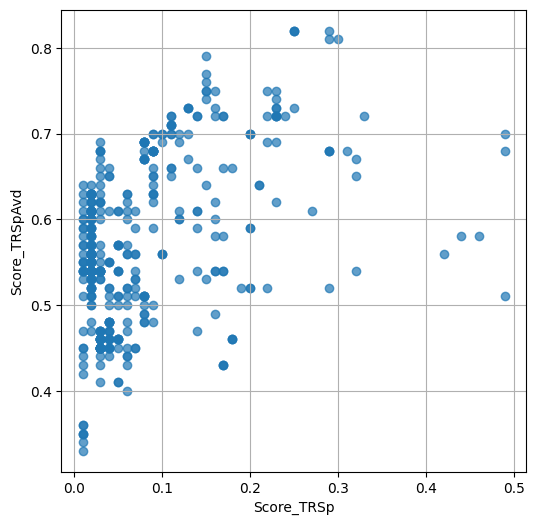
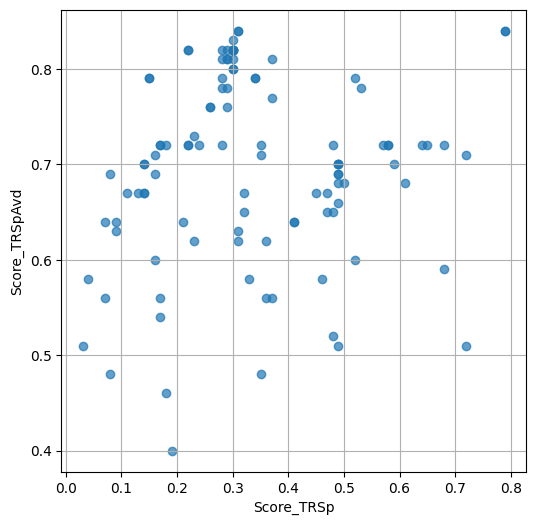
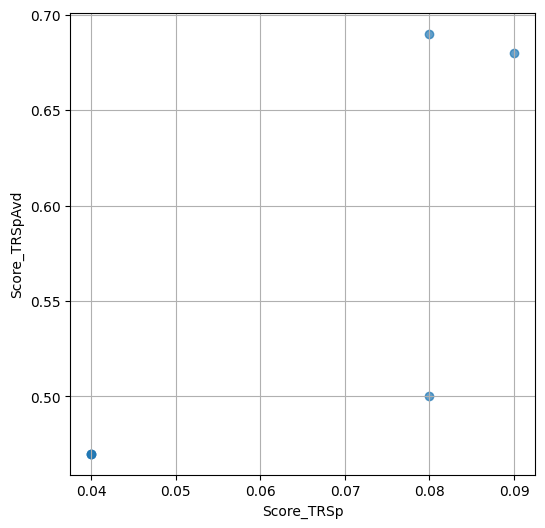
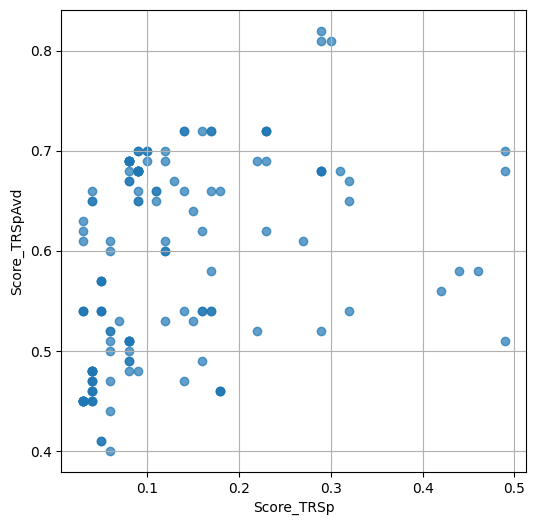
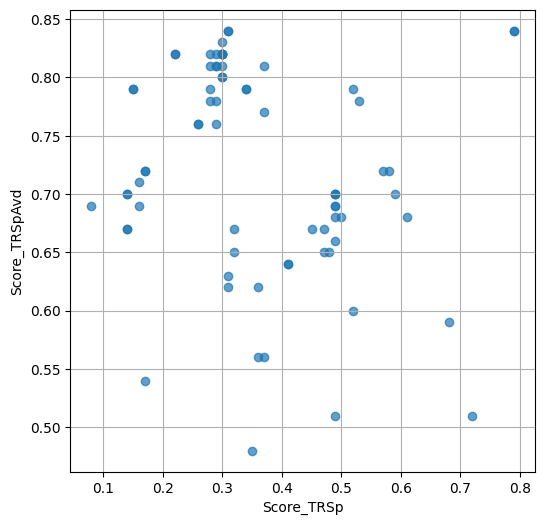
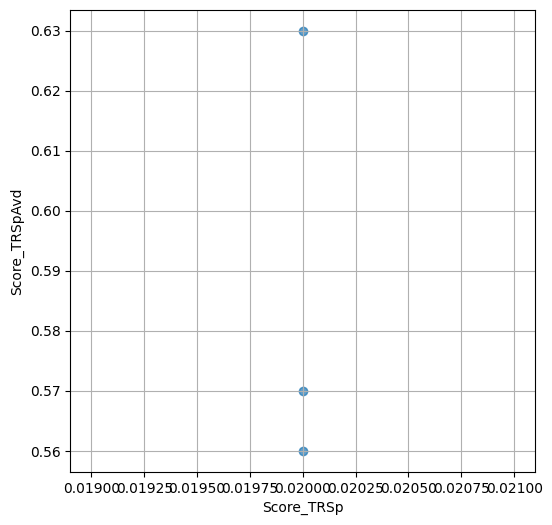
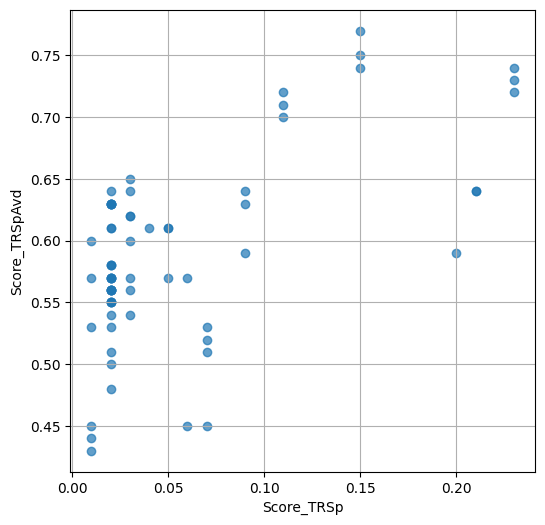
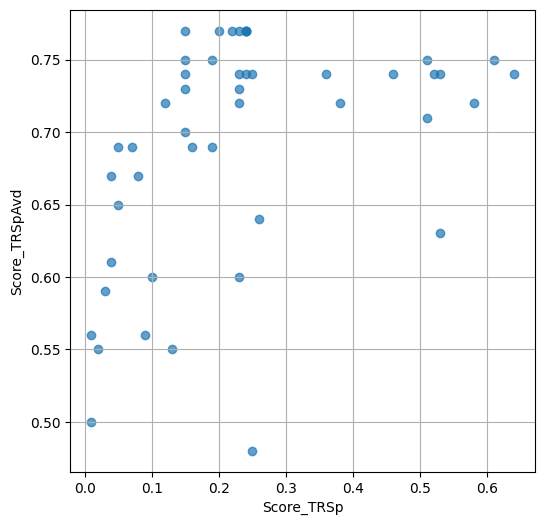
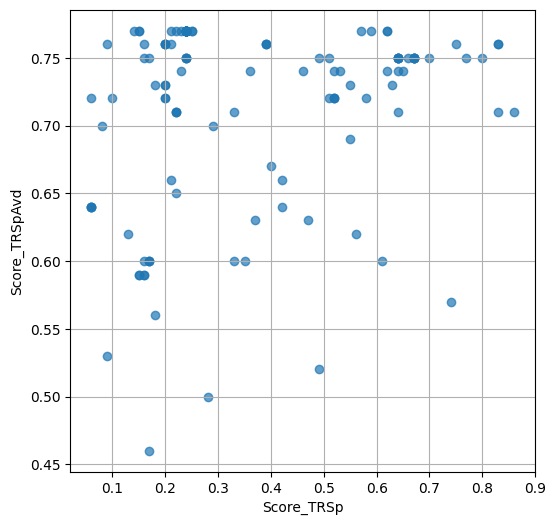
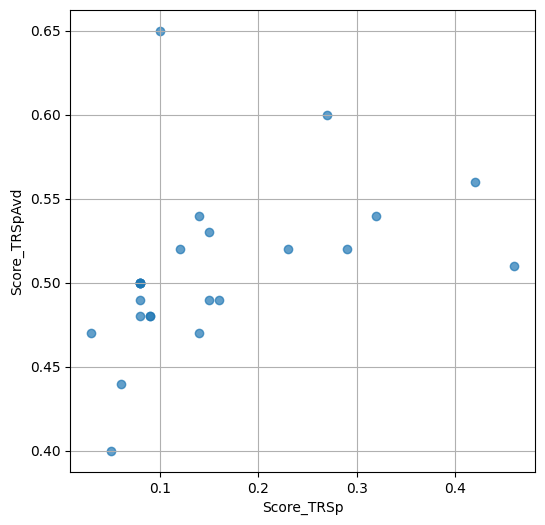
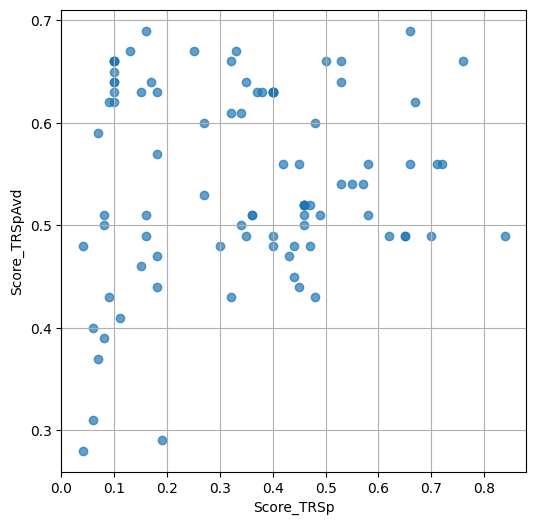
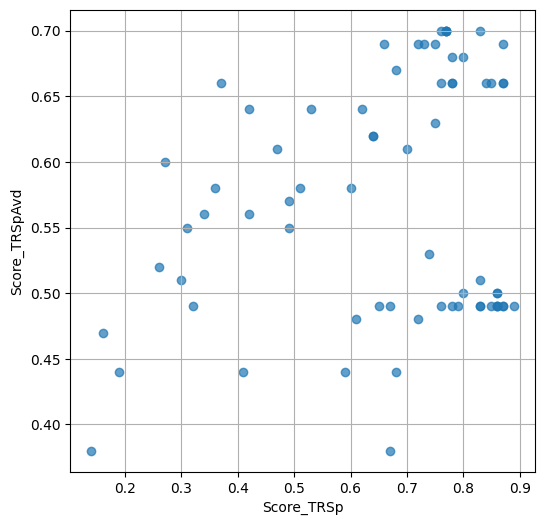
threshold:float=0.7, # Minimum required value for both `Score_TRSp` and `Score_TRSpAvd` to
accept a sequence as optimal.
codon_usage:dict={'F': {'TTT': 0.57, 'TTC': 0.43}, 'L': {'TTA': 0.15, 'TTG': 0.12, 'CTT': 0.12, 'CTC': 0.1, 'CTA': 0.05, 'CTG': 0.46}, 'S': {'TCT': 0.11, 'TCC': 0.11, 'TCA': 0.15, 'TCG': 0.16, 'AGT': 0.14, 'AGC': 0.33}, 'Y': {'TAT': 0.53, 'TAC': 0.47}, '*': {'TAA': 0.64, 'TAG': 0.0, 'TGA': 0.36}, 'C': {'TGT': 0.42, 'TGC': 0.58}, 'W': {'TGG': 1.0}, 'P': {'CCT': 0.17, 'CCC': 0.13, 'CCA': 0.14, 'CCG': 0.55}, 'H': {'CAT': 0.55, 'CAC': 0.45}, 'Q': {'CAA': 0.3, 'CAG': 0.7}, 'R': {'CGT': 0.36, 'CGC': 0.44, 'CGA': 0.07, 'CGG': 0.07, 'AGA': 0.07, 'AGG': 0.0}, 'I': {'ATT': 0.58, 'ATC': 0.35, 'ATA': 0.07}, 'M': {'ATG': 1.0}, 'T': {'ACT': 0.16, 'ACC': 0.47, 'ACA': 0.13, 'ACG': 0.24}, 'N': {'AAT': 0.47, 'AAC': 0.53}, 'K': {'AAA': 0.73, 'AAG': 0.27}, 'V': {'GTT': 0.25, 'GTC': 0.18, 'GTA': 0.17, 'GTG': 0.4}, 'A': {'GCT': 0.11, 'GCC': 0.31, 'GCA': 0.2, 'GCG': 0.38}, 'D': {'GAT': 0.65, 'GAC': 0.35}, 'E': {'GAA': 0.7, 'GAG': 0.3}, 'G': {'GGT': 0.29, 'GGC': 0.46, 'GGA': 0.13, 'GGG': 0.12}}, # Codon usage table of E. Coli mapping amino acids to codons and frequencies.
): # Dictionary containing:
- `Original_Sequence` : str
Input DNA sequence.
- `New_Variant` : str
Optimized DNA sequence.
- `Rank` : int or None
rank of the sequence (by score).
- `Score` : float or None
score of the selected variant (geometrical mean).
- `Score_TRSp` : float or None
TR+Sp score of the selected variant.
- `Score_TRSpAvd` : float or None
Avd+TR+Sp score of the selected variant.